| 1.必须从受试者或其法定代表处(如使用)获得经机构审查委员 会(IRB)/独立伦理委员会(IEC)根据国家法规(例如美国研究 中心的健康保险流通与责任法案[HIPAA]授权)批准的书面知情同 意书和保密信息。 |
| 2.在签署知情同意书时,受试者根据当地法规被视为成人(例如 在美国,年龄为≥ 18 岁)。 |
| 3.女性受试者如果符合以下条件就有资格参加研究:未怀孕(筛 选时血清妊娠试验阴性;血清 β 人绒毛膜促性腺激素[β-hCG] 升高但通过其他检查证明不是妊娠状态的女性受试者有资格参加 研究)并且适用以下至少 1 个条件: ● 不是[附录 12.3 避孕 要求]中定义的有生育能力女性(WOCBP) 或者 ● 是 WOCBP 但 同意在整个治疗期间以及至研究治疗末次给药后 6 个月遵守[附 录 12.3 避孕要求]定义的避孕指导 |
| 4.女性受试者必须同意在整个研究期内自筛选时起开始停止哺乳 并持续至末次研究药物给药后 6 个月。 |
| 5.女性受试者在整个研究期内自筛选时起不得捐赠卵子并持续至 末次研究药物给药后 6 个月。 |
| 6.女性伴侣有生育能力的男性受试者必须同意在治疗期间以及至 研究治疗末次给药后6个月采用[附录12.3避孕要求]要求的避孕 措施。 |
| 7.男性受试者必须同意从筛选开始,在整个研究期间以及至药物 末次给药后 6 个月内不得捐精。 |
| 8.整个研究期间以及研究药物末次给药后 6 个月内,有妊娠或哺 乳伴侣的男性受试者必须同意在其伴侣妊娠期间或哺乳期间禁欲 或使用避孕套。 |
| 9.受试者同意在本研究中接受研究药物时不会参加另一项干预性 研究。 |
| 10.受试者经组织学证实诊断为胃或 GEJ 腺癌。 |
| 11.受试者在研究治疗首次给药前 28 天内经影像学证实为局部晚 期不可切除或转移性疾病。 |
| 12.受试者在研究治疗首次给药前 28 天内根据 RECIST 1.1 有可测 量的疾病。对于只有 1 个可测量病灶和接受既往放疗的受试者, 病灶必须在既往放疗范围外,或必须在放疗后有记录的进展。 |
| 13.受试者的肿瘤有≥ 75%肿瘤细胞表达 CLDN18.2,中心实验室 IHC 检测确定有中至重度膜染色。 |
| 14.受试者患有已知 HER2 阴性的胃或 GEJ 肿瘤。 |
| 15.受试者的 ECOG 体能状态为 0 或 1。 |
| 16.研究者认为受试者的预期寿命≥12 周。 |
| 17.在研究治疗首次给药之前 14 天内,根据中心实验室的检查分 析,受试者必须符合以下所有标准。在筛选期内有多个中心实验 室数据的情况下,应采用最近的数据确定资格。 ● 血红蛋白 (Hgb)≥ 9 g/dl。注意:受试者在筛选时获得血液学数值前 14 天内不得接受任何生长因子或输血。需要输血才能符合参试标准 的受试者没有参试资格。 ● 中性粒细胞绝对计数(ANC) ≥1.5 × 109/L ● 血小板≥ 100 × 109/L ● 白 蛋白≥2.5 g/dL ● 总胆红素≤1.5 倍正常上限(ULN) ● 天冬氨酸氨基转移酶 (AST) 和丙氨酸氨基转移酶 (ALT) ≤2.5 倍 ULN,并且无肝转移(若发生肝转移,则为 ≤5 倍 ULN)。 ● 估计的肌酐清除率≥ 30 mL/min ● 凝 血酶原时间/国际标准化比值(INR)和部分凝血活酶时间 (PTT)≤1.5 倍 ULN(除接受抗凝治疗的受试者外) |
*以上临床试验信息摘自国家药品监督管理局药品审评中心临床试验登记与信息公示平台,具体入排以研究中心医生评估为准*
| 1.受试者既往已接受了全身性化疗治疗局部晚期不可切除或转移 性胃或 GEJ 腺癌。但是只要受试者在研究治疗首次给药前至少 6 个月已完成新辅助化疗或辅助化疗的则可以接受。 |
| 2. 受试者已接受了局部晚期不可切除或转移性胃癌或 GEJ 腺癌的 放疗治疗,除非放疗在研究治疗首次给药前>28 天已完成。受试 者在研究治疗首次给药前≥ 14 天接受周围骨转移的姑息放疗治 疗并已从所有急性毒性中恢复,则允许。 |
| 3. 受试者研究治疗首次给药前 28 天内已接受已知的具有抗肿瘤 活性的草药或其他治疗。 |
| 4. 受试者在研究治疗首次给药前 14 天内已接受了全身性免疫抑 制疗法,包括全身性皮质类固醇治疗。允许受试者使用生理替代 剂量的氢化可的松或其等价物(定义为氢化可的松每日最高 30 mg 或强的松每日最高 10 mg),或者接受单次全身性皮质类固醇治 疗。 |
| 5. 受试者在研究治疗首次给药前 28 天内已接受了其他研究药物 或器械。 |
| 6. 受试者有既往严重过敏反应或对 Zolbetuximab 的已知组分或 其他单克隆抗体(包括人源化或嵌合抗体)不耐受。 |
| 7. 受试者已知对研究治疗的任何成分有立即或延迟的超敏反应、 不耐受或禁忌。 |
| 8. 受试者既往对 CAPOX 的任何成分有严重过敏反应或不耐受。 |
| 9. 受试者已知有二氢嘧啶脱氢酶(DPD)缺乏症(注意:应根据当 地要求进行 DPD 缺乏症筛查。) |
| 10. 受试者患有胃出口综合征或持续/反复呕吐。 |
| 11. 最近出现胃出血的受试者或有证据表明胃溃疡的有症状受试 者,根据研究者的判断将排除受试者的参与。 |
| 12. 受试者有已知的人类免疫缺陷病毒(HIV)感染或已知的活动 性乙型肝炎(阳性 HBsAg)或丙型肝炎感染的阳性检测史。对于 HBsAg 阴性但 HBc Ab 阳性的受试者,将进行 HBV DNA 检测,如果 呈阳性则排除受试者。血清学阳性但丙型肝炎病毒(HCV)RNA 检 测结果阴性的受试者具有入组资格。 |
| 13. 受试者过去 2 年内患有需要全身性治疗的活动性自身免疫性 疾病。 |
| 14. 受试者患有需要全身性治疗的活动性感染且在研究治疗首次 给药前 14 天内未完全缓解。 |
| 15. 受试者患有重大心血管疾病,包括下列任何情况: 1. 在 第一剂研究药物给药前 6 个月内的充血性心力衰竭(定义为纽约 心脏协会[NYHA]III 或 IV 级)、心肌梗塞、不稳定型心绞痛、冠 状动脉血管成形术、支架置入术、冠状动脉搭桥术、脑血管意外 (CVA)或高血压危象; 2. 有临床意义的室性心律失常病史(如 持续性室性心动过速,室颤,尖端扭转型室性心动过速); 3. 男性受试者:QTc 间期> 450 毫秒;女性受试者:QTc 间期> 470 毫秒; 4. 先天性长 QT 综合征病史或家族史; 5. 需要抗心律 失常药物治疗的心律失常(在研究药物首次给药之前> 1 个月患 有心率可控制的房颤的受试者有入组资格) |
| 16. 受试者已知有中枢神经系统(CNS)转移和/或癌性脑膜炎。 |
| 17. 受试者已知患有> 1 级的外周感觉神经病变,除非深部肌腱反 射缺失是唯一的神经异常。 |
| 18. 受试者在研究治疗首次给药前≤28 天已接受了重大手术。 a) 受试者在研究治疗首次给药前≤14 天未从重大手术中康复。 |
| 19. 根据研究者的判断,受试者患有会妨碍研究依从性的精神疾病 或社交情况。 |
| 20. 根据研究者的判断,受试者患有会妨碍研究依从性的精神疾病 或社交情况。 |
| 21. 根据研究者的判断,受试者患有会妨碍研究依从性的精神疾病 或社交情况。 |
*以上临床试验信息摘自国家药品监督管理局药品审评中心临床试验登记与信息公示平台,具体入排以研究中心医生评估为准*








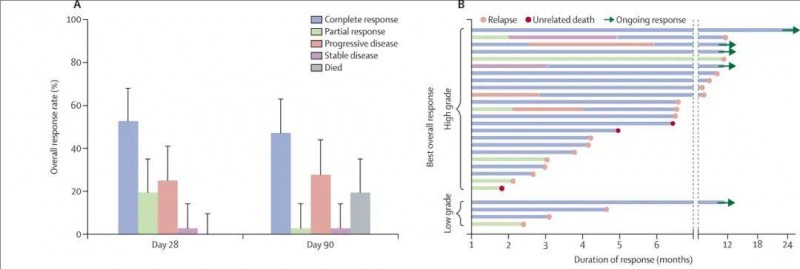
![[field:title/]](/uploads/allimg/20200224/1-2002241I5262J.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241I3461D.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H94LK.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241HKW34.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H619327.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H34H52.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H22IH.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241G93O18.jpg)




