| 1 | 准备进行或者既往接受过器官或骨髓移植的受试者。 |
| 2 | 通过适当干预后无法控制的胸腔积液、心包积液或腹水。 |
| 3 | 已知或筛选期检查发现患有活动性中枢神经系统转移和/或癌性脑膜炎者。但允 许以下受试者入组: 1. 无症状性脑转移受试者(即没有脑转移灶引起的进行性 中枢神经系统症状,不需要皮质类固醇,且病变大小≤ 1.5cm)可以参加,但需 要作为疾病部位定期进行脑部影像学检查。 2.经治疗后的脑转移受试者,且脑转 移病灶稳定至少 1 个月,没有新的或扩大的脑转移证据,且本研究药物首次给 药前 3 天停用类固醇。这个定义中稳定的脑转移应在研究药物首次给药前确定。 |
| 4 | 手术和/或放疗未能根治性治疗的脊髓压迫,或既往诊断的脊髓压迫经治疗后没 有临床证据显示在研究药物首次用药前疾病稳定 ≥ 1 周的受试者。 |
| 5 | 影像学检查显示,存在明确的肿瘤侵犯胸部大血管表现。 |
| 6 | 患有Ⅱ级以上心肌缺血或研究药物首次用药前半年内发生过心肌梗塞、 不稳定心 绞痛、 控制不良的心律失常(包括 QTc 间期男性 ≥ 450 ms、女性 ≥ 470 ms)(QTc 间期以 Fridericia 公式计算)。 |
| 7 | 按照美国纽约心脏病学会(NYHA) 标准(附录三) Ⅲ级或Ⅳ级心功能不全或心 脏彩超检查: 左室射血分数(LVEF) < 50%。 |
| 8 | 存在 CTCAE v4.03 ≥ 2 级的外周神经病变。 |
| 9 | 存在 CTCAE v4.03 ≥ 2 级的外周神经病变。 |
| 10 | 患有活动性肺结核病。 |
| 11 | 既往或目前有间质性肺炎、尘肺、放射性肺炎、药物相关肺炎、肺功能严重受损 等可能会干扰可疑的药物相关肺毒性的检测和处理。 |
| 12 | 存在已知活动性或可疑自身免疫病。允许入组处于稳定状态且不需要全身免疫抑 制剂治疗者。 |
| 13 | 研究药物首次用药前 28 天内接受过活疫苗的治疗。 |
| 14 | 研究药物首次用药前 14 天内或研究期间, 需要全身使用皮质类固醇(> 10 mg/天 泼尼松或等效剂量的同类药物)或其他免疫抑制剂治疗的受试者。 但以下情况允 许入组: 在没有活动性自身免疫疾病的情况下,允许吸入, 或局部外用类固醇, 或剂量≤ 10 mg/天泼尼松疗效剂量的肾上腺激素替代治疗。 |
| 15 | 研究药物首次用药前 14 天内,出现任何需要系统性给予抗感染治疗的活动性感 染。 |
| 16 | 研究药物首次用药前 28 天内,接受过重大手术,本研究重大手术定义:术后至 少需要 3 周恢复时间,才能够接受本研究治疗的手术。 肿瘤穿刺或淋巴结切取活 检允许入组。 |
| 17 | 研究药物首次用药前 3 个月内,接受过根治性放射治疗。 注:允许接受骨的姑息性放疗或浅表病灶的姑息性放疗,疗程参照当地的标准, 且在首次用药前 14 天已经结束。不允许首次给药前 28 天内接受覆盖 30%以上 骨髓区域的放疗。 |
| 18 | 在研究期间可能会接受其他抗肿瘤治疗,如化疗、靶向治疗或放疗(姑息性放疗 除外)。 |
| 19 | 既往接受过任何 T 细胞共刺激或免疫检查点治疗,包括但不限于细胞毒性 T 淋 巴细胞相关抗原-4(cytotoxic T lymphocyte-associated antigen-4, CTLA-4)抑制剂、 PD-1 抑制剂、 PD-L1/2 抑制剂或其他靶向 T 细胞的药物。 |
| 20 | 正在参加其他临床研究,或计划开始本研究治疗距离前一项临床研究药物治疗结 束时间不足 14 天。 |
| 21 | 已知对任何单克隆抗体或研究药物辅料有严重过敏史。 |
| 22 | 妊娠期或哺乳期女性。 |
| 23 | 已知有精神类药物滥用或吸毒史。已停止饮酒的受试者可以入组。 |
| 24 | 经研究者判断,受试者具有其他可能导致本研究被迫中途终止的因素。 |








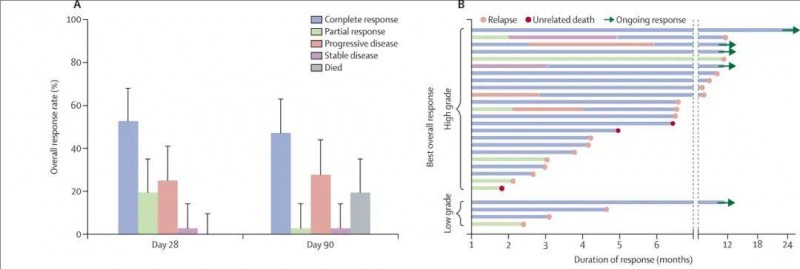
![[field:title/]](/uploads/allimg/20200224/1-2002241I5262J.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241I3461D.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H94LK.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241HKW34.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H619327.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H34H52.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H22IH.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241G93O18.jpg)




