



【临床试验招募】Tepotinib临床试验
招募标准
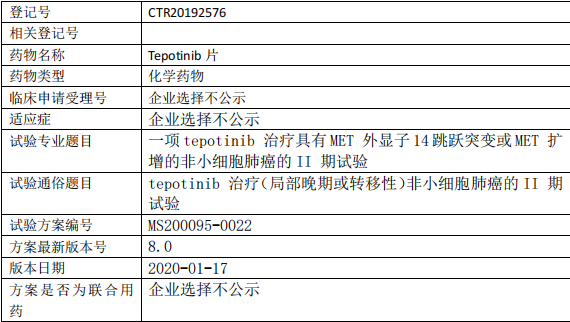
Tepotinib临床试验,Tepotinib片治疗(局部晚期或转移性)非小细胞肺癌的II期试验
1、试验目的
主要目的:
第 1 部分:队列 A(METex14 跳跃突变): 基于对以下患者的独立审查,评估 tepotinib 在晚期(局部晚期或转移性)非小细胞肺癌受试者中的有效性(根据实体瘤疗效评价标准[RECIST]1.1 版确定的客观缓解(经确认的完全缓解[CR]或部分缓解[PR])
第 1 部分:队列 B(MET 扩增): 基于对以下患者的独立审查,评估 tepotinib 在晚期(局部晚期或转移性)非小细胞肺癌受试者中的有效性(根据 RECIST 1.1 版确定的客观缓解[经确认的 CR 或 PR])
第 2 部分:队列 C(METex14 跳跃突变的确证性部分) 基于对以下患者的独立审查,评估 tepotinib 在晚期(局部晚期或转移性)非小细胞肺癌受试者中的有效性(根据 RECIST 1.1 版确定的客观缓解[经确认的 CR 或 PR])
2、试验设计
试验分类:安全性和有效性
试验分期:II 期
设计类型:单臂试验
随机化:非随机化
盲法:开放
试验范围:国际多中心试验
3、受试者信息
年龄:18 岁(最小年龄)至无最大年龄岁(最大年龄)
性别:男+女
健康受试者:无
入排标准
入选标准
1.在任何试验特异性筛选程序之前,受试者或其法定代理人签署了知情同意书;
2.男性或女性,年龄≥18 岁(或者如果根据当地法律和法规的成年年龄>18 岁,则需要达到成年年龄[例如,在日本应≥20 岁])
3.通过 IRC 确认疾病可测量(根据 RECIST 1.1 版);
4.东部肿瘤协作组体能状态(ECOG PS)评分为 0 或 1;
5.女性受试者如果不在妊娠期,不在哺乳期,而且至少符合以下一种情况,则可以参加研究:
a. 不属于附录 VIII 中定义的具有生育能力的女性 或者
b.育龄期女性必须同意从研究治疗第一次给药开始前 2 周至治疗期间以及研究治 疗最后一次给药后至少 4 周使用本研究方案附录 VIII 中详述的高效避孕方法 (即,每年失败率低于 1%的方法)。育龄期女性在入组前的妊娠试验(血清β-HCG 检测)结果必须为阴性。
6.男性受试者必须同意从研究治疗第一次给药开始至治疗期间以及研究治疗最后一次给药后至少 3 个月内使用本研究方案附录VIII 中详述的高效避孕方法(即,每年 失败率低于 1%的方法),他们的育龄女性伴侣也必须使用,并且在此期间不得捐献精子。男性受试者应始终同时使用避孕套等屏障方法。
7.经组织学或细胞学证实的晚期(局部晚期或转移性)NSCLC(所有类型,包括鳞 状和肉瘤样);
8.未经治疗的一线患者,或接受过不超过 2 线既往治疗的经治疗患者
9.受试者存在 MET 突变,即
1)血浆和/或组织中存在 METex14 跳跃突变(通过中心实验室或通过具有适当监 管状态的测定法来确定)的受试者将入组本试验。对于这些受试者,需要足够的肿瘤组织和/或血浆来进行额外的检测
2)仅血浆中MET扩增定义为LBx检测结果阳性(通过中心实验室或通过具有适当监管状态的测定法来确定)
3)基于对 12 例 LBx 选定受试者的中期分析结果:仅组织中 MET 扩增定义为 TBx 检 测结果阳性,MET 基因拷贝数增加至少 4 个(通过中心实验室或通过具有适当监管状态的测定法来确定)。
排除标准
排除标准
1.有症状脑转移的受试者神经系统不稳定,和/或需要增加在 2 周内增加类固醇剂量和/或在 2 周内接受既往立体定向放射外科/伽玛刀治疗,和/或在开始治疗前 4 周内接受其他既往脑转移治疗。患有软脑膜疾病的受试者不符合要求;
2. 根据国家癌症研究所-不良事件通用术语标准(NCI-CTCAE)4.03版,来自以前抗 癌治疗的任何未痊愈的≥2 级毒性;
3. 需要在试验治疗第一次给药前 14 天内进行输血。
4. 脑转移是唯一可测量病灶的受试者。
5. 具有预测抗 EGFR 治疗敏感性的 EGFR 激活突变特征的受试者;
6. 具有预测抗 ALK 治疗敏感性的 ALK 重排特征的受试者;
7. 在试验治疗第一次给药前 21 天内以抗肿瘤为目的的既往化疗、生物治疗、放射治疗、激素治疗、靶向治疗或其它试验性抗癌治疗(不包括病灶部位的姑息性放射治疗)。
8. 实验室检查值和器官功能,血液系统功能不全:血红蛋白< 8.5g/dL;中性粒细胞< 1.5 × 109/L;血小板< 100 × 109/L。
9. 肝功能不全:总胆红素>1.5 x 正常值范围上限(ULN);天冬氨酸氨基转移酶 (AST)/丙氨酸氨基转移酶(ALT) 3×ULN;对于有肝转移的受试者:总胆红素> 1.5 × ULN,AST/ALT > 5 × ULN
10. 肾功能不全:重度肾损害,表现为:血清肌酐≥ 1.5 × ULN;肌酐清除率(CrCl) < 30 mL/min,通过 Cockcroft-Gault 公式计算(如果计算得到 的 CrCl < 30 mL/min,研究者可能会要求24小时 CrCl 以进行确认)。在这种情况下, 应排除 24 小时CRCl<30 mL/min 的受试者)CrCl (mL/min) = [140 – 年龄(岁)× 体重(kg)] 72 × 血清肌酐(mg/dL) {× 0.85,对于女性受试者}
11. 一般标准,既往接受靶向 HGF/c-Met 通路的其它药物进行治疗;
12. 心功能受损
a.超声心动图确定的左心室射血分数<45%(除非有临床指征,否则不需要对没 有充血性心力衰竭病史的受试者进行筛选评估)
b.严重心律失常
c.不稳定型心绞痛
d.纽约心脏学会III 级和 IV 级心力衰竭
e.试验开始前 12 个月内发生过心肌梗死
f.症状性心包积液
13. 标准治疗无法控制的高血压(未稳定至 150/90 mmHg)
14. NSCLC 以外肿瘤的病史或现病史,不包括经根治性治疗的非黑色素瘤皮肤癌、宫 颈原位癌,或者其他经过根治性治疗且至少 5 年无疾病迹象的癌症
15. 吞咽困难、吸收不良或其它慢性胃肠道疾病病史,或可能妨碍测试产品依从性和/ 或吸收的病症
16. 第 1 天试验治疗前 28 天内接受过大手术
17. 已知感染人免疫缺陷病毒,或活动性乙型肝炎或丙型肝炎病毒感染
18. 药物滥用、活动性感染或其它急性或慢性医学或精神疾病,或根据研究者的判断 可能会增加参与试验风险的实验室异常
19. 已知对任何试验治疗成分存在超敏反应
20. 已知对任何试验治疗成分存在超敏反应
21. 无民事行为能力或限制民事行为能力22. 被主要研究者认为会妨碍受试者参加试验的任何其它原因
23. 过去 30 天内参与过另一项临床试验。
研究者信息
1、主要研究者信息
姓名:吴一龙
学位:医学博士
职称:主任医师
单位名称:广东省人民医院
2、各参加机构信息
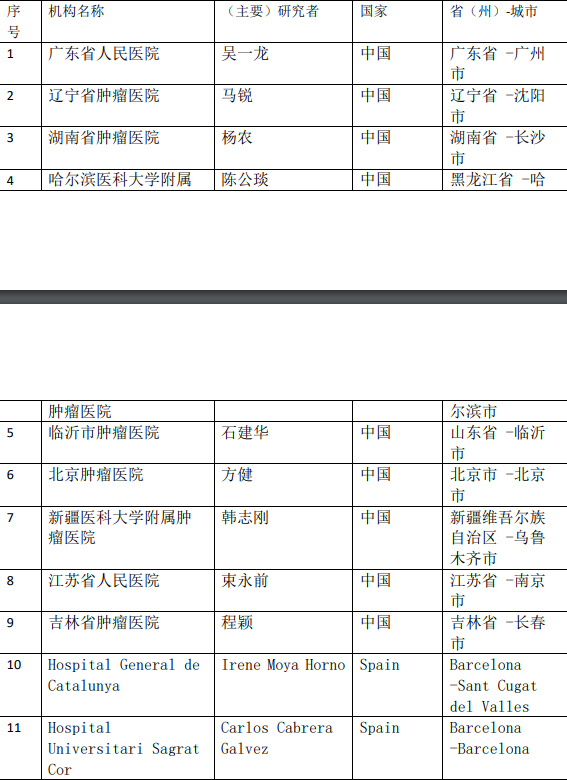
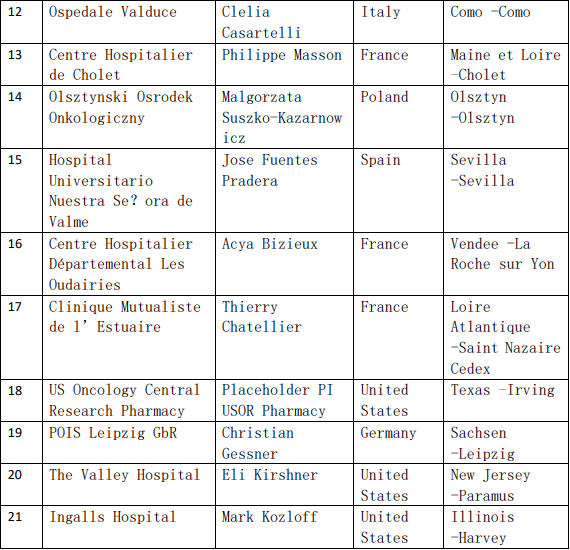
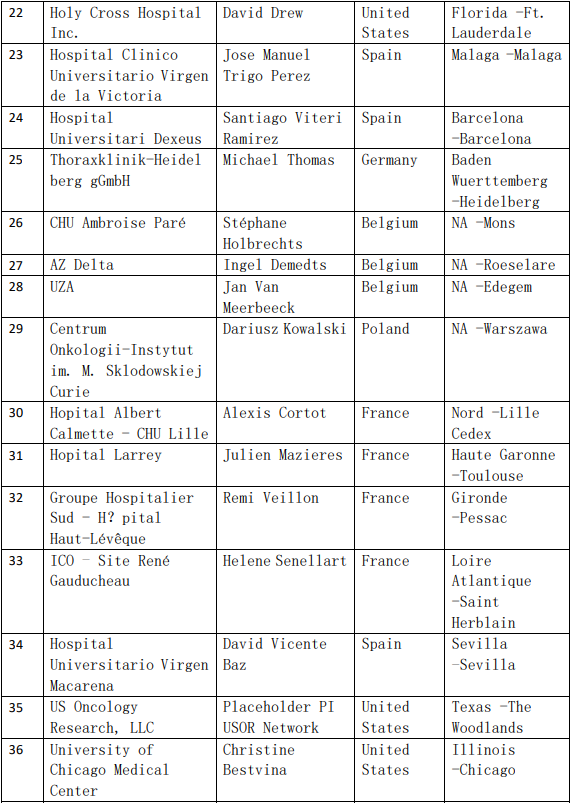
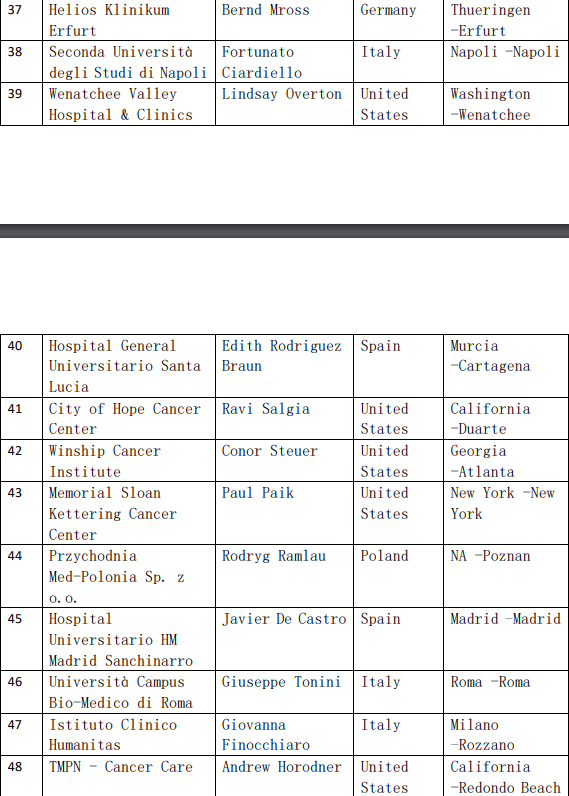
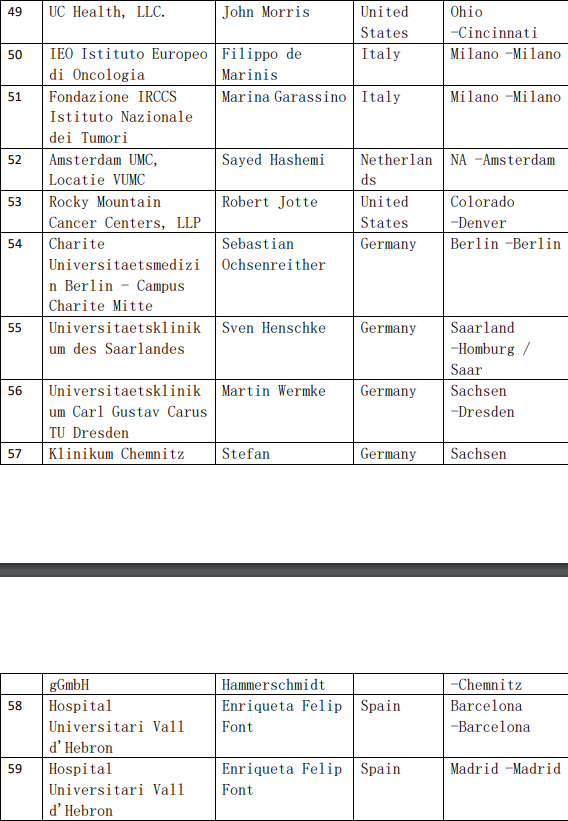
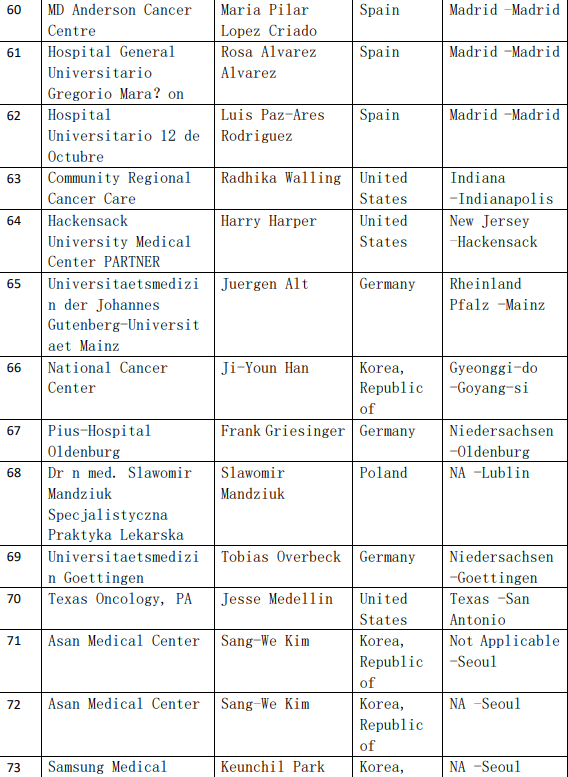
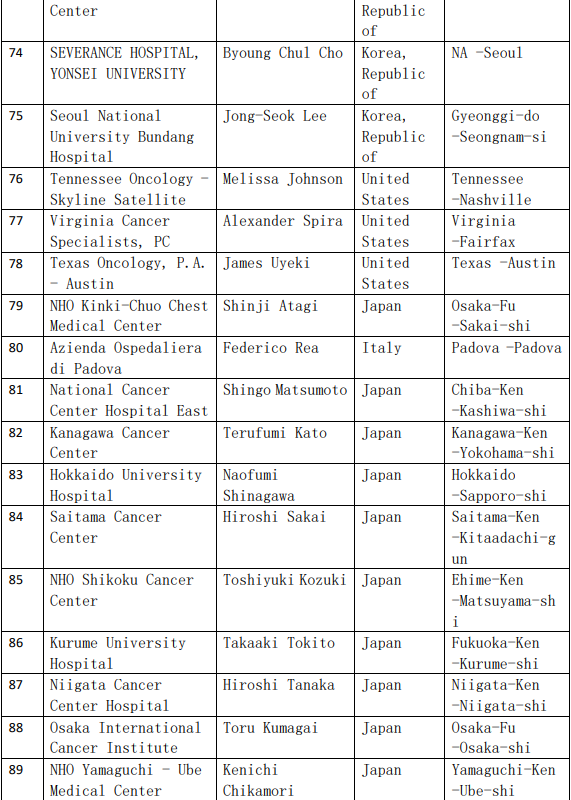
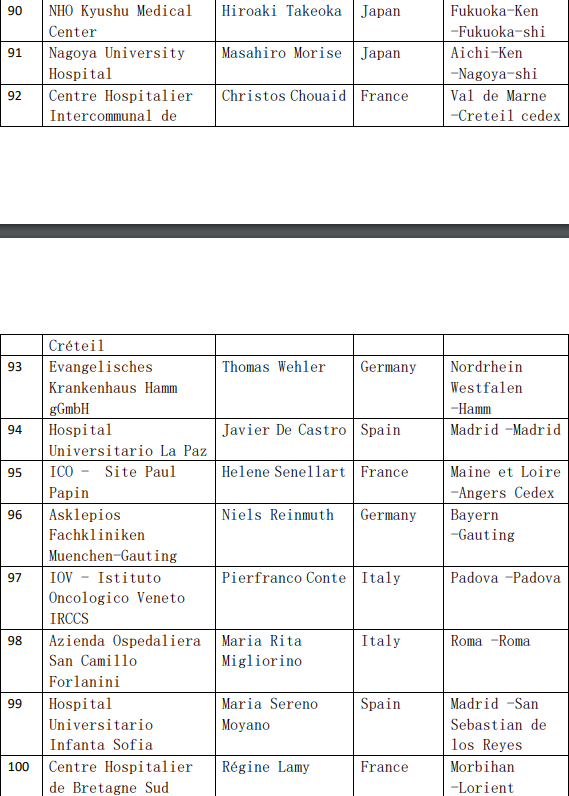
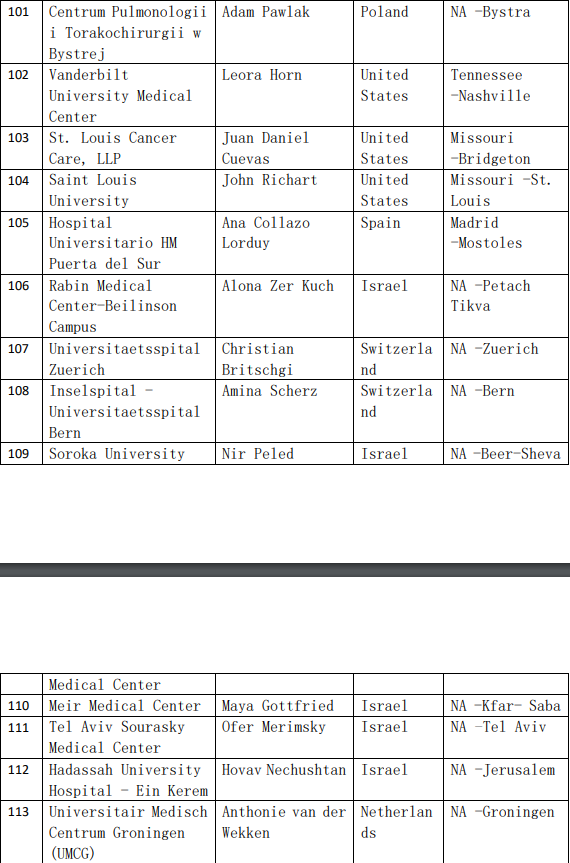
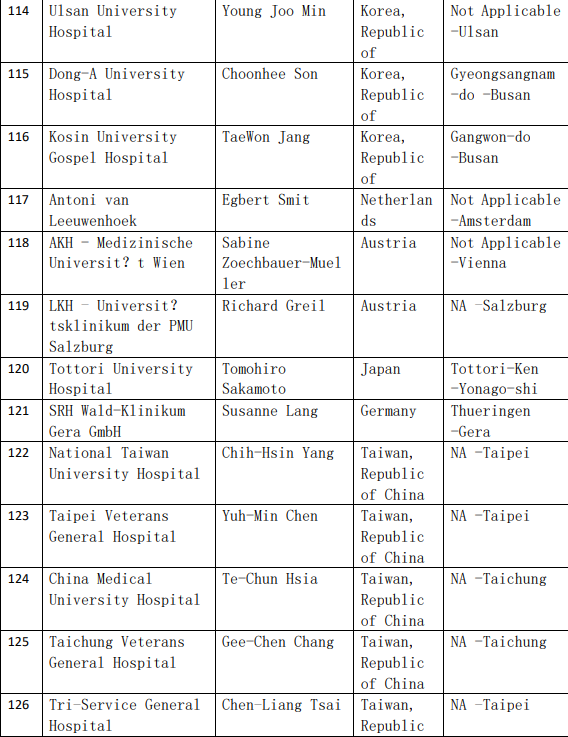
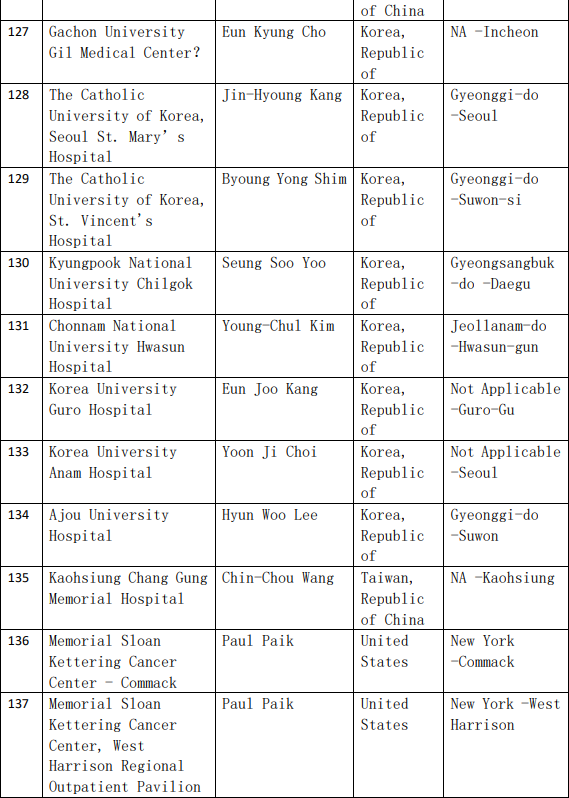
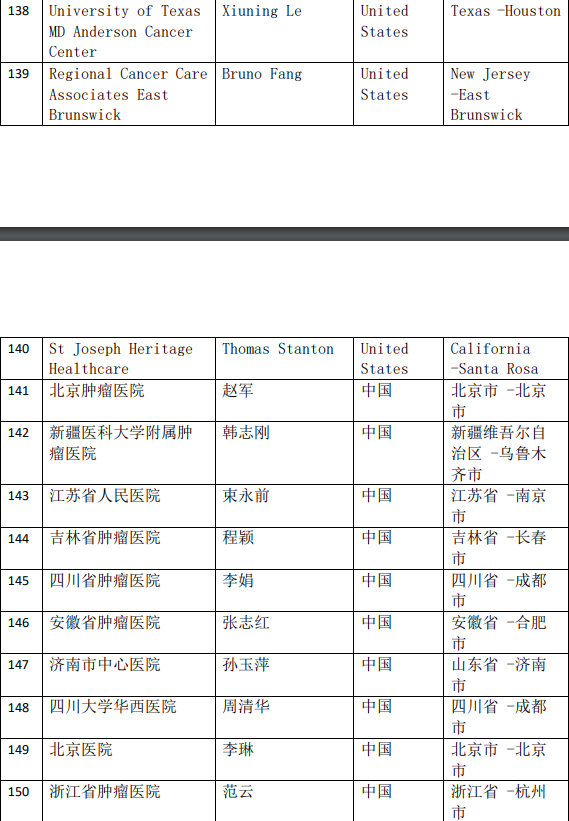







![[field:title/]](/uploads/allimg/20200224/1-2002241I5262J.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241I3461D.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H94LK.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241HKW34.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H619327.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H34H52.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241H22IH.jpg)
![[field:title/]](/uploads/allimg/20200224/1-2002241G93O18.jpg)




